The long read mapper ALFALFA achieves high performance in accurately mapping long (>500bp) single-end and paired-end reads to gigabase-scale reference genomes, while remaining competitive for mapping shorter (>100bp) reads. Its seed-and-extend workflow is underpinned by fast retrieval of super-maximal exact matches from an enhanced sparse suffix array, with flexible parameter tuning to balance performance, memory footprint and accuracy.
The following commands can be used to install the software if git is installed.
The following commands can be used to unpack and install the software when downloading a zip file. Replace VERSION with the name of the downloaded file.
The following commands can be used to unpack and install the software when downloading a tarball. Replace VERSION with the name of the downloaded file.
Options can have a single-letter (preceded by a single hyphen) or multi-letter (preceded by a double hyphen) name, or both. In the latter case, both names of the option can be used interchangeably. The description of an option starts with its name or names (separated by a forward slash), followed by a tuple (between round brackets) indicating the data type and default value of the argument that has to be passed to the option. If no default value is given, it is mandatory to pass an argument to the option. Options for which no tuple is given are used as toggles to enable/disable certain features and need no extra argument.
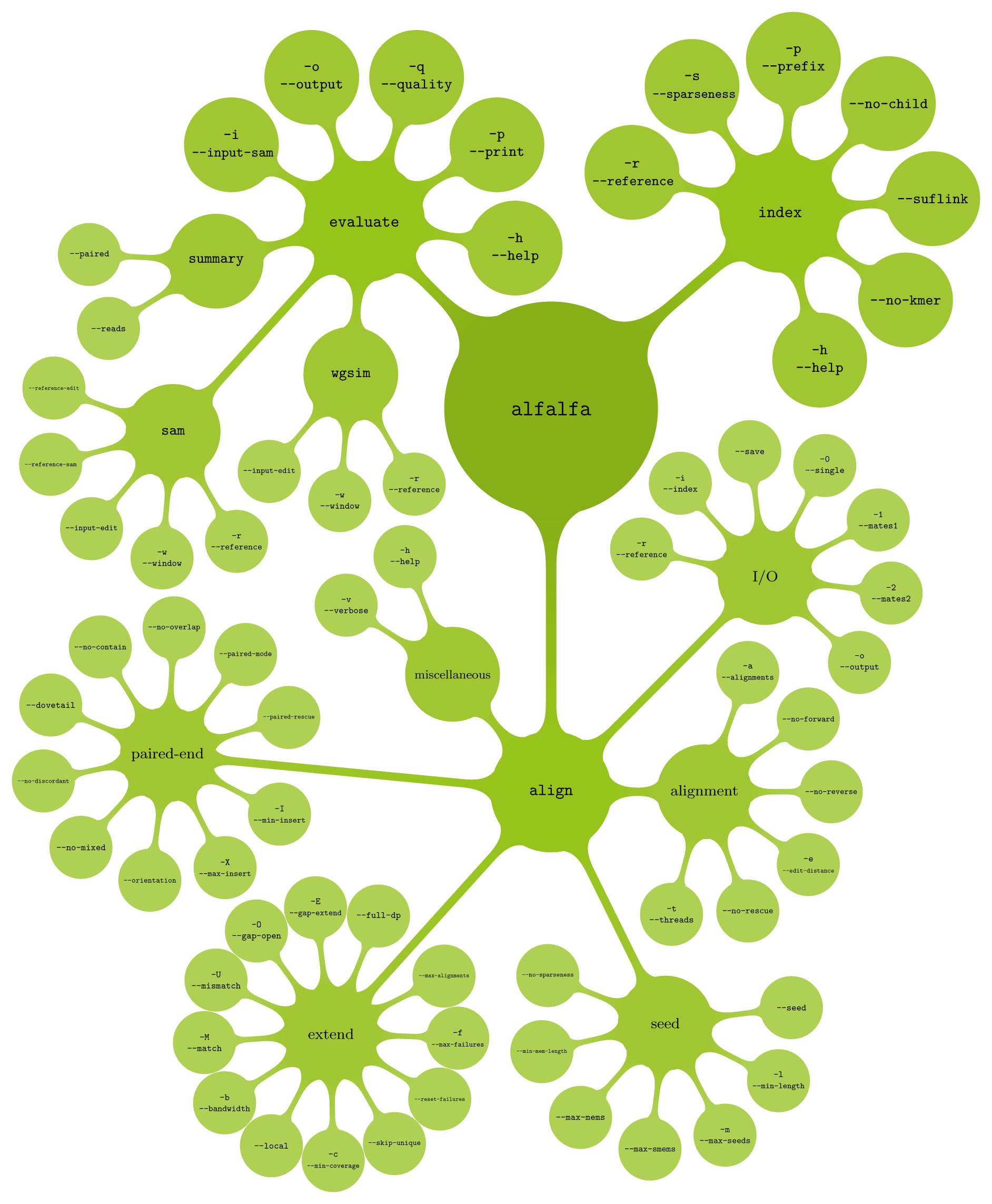
The following graphical overview of the anatomy of ALFALFA provides details on the command line options that can be used to tweak its operation. The software package offers separate commands for index construction, read mapping and evaluating mapping accuracy. Index construction can also be combined with read mapping during a single run of the package. Click on green regions to get a detailed description and general usage tips of each command, subcommand and option supported by ALFALFA. Click on white background to get a full screen display of the command line anatomy of ALFALFA.
To quickly get started, you can use the small example files contained within the repository and the commands given in the example page.
Contact lead developer Michaël Vyverman (michael[dot]vyverman[at]ugent[dot]be) if you have any further questions or suggestions.